Keywords
COVID-19; SARS-CoV-2, repurposed drugs, RCT, adaptive design, early treatment, outpatient care, master protocol
This article is included in the Coronavirus (COVID-19) collection.
COVID-19; SARS-CoV-2, repurposed drugs, RCT, adaptive design, early treatment, outpatient care, master protocol
The discovery of effective and affordable treatments for preventing COVID-19 disease progression and subsequent hospitalization in outpatient settings is critical to minimize limited hospital resources, particularly for resource-limited settings1. As vaccine rollout has been slow in resource-limited countries and new variants of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) cause concern for their effectiveness, identifying therapeutics that are affordable, widely available and effective against COVID-19 is of prime importance. Repurposing existing medications is an appealing approach as drugs currently used to treat other health conditions have known safety profiles.
There is also a need for more clinical trials in early infected populations. A majority of trials of repurposed drugs are conducted among inpatients with advanced clinical disease, yet the majority of COVID-19 cases are seen in the community setting2. Early treatment trials have the added benefit of evaluating drugs with the outcome of disease progression or hospitalization3. The TOGETHER Trial is an example of a global study network to evaluate repurposed drugs in early infected populations.
The TOGETHER Trial is an adaptive, multi-arm platform trial, evaluating multiple concurrent interventions (investigational products [IPs]) versus placebo among outpatients at high risk of developing COVID-19-related complications. The trial is designed to allow for multiple intervention arms to be implemented at any time and data to be merged with data from other external trials. This is a new approach for clinical trials that has occurred as a result of the COVID-19 pandemic and integrates platform adaptive trial designs with data synthesis to facilitate rapid decision-making. The overarching objective of this study is to test the hypothesis that repurposed drugs versus placebo effectively prevent worsening of COVID-19 requiring hospitalization or emergency room observation for greater than 6 hours among high-risk adults at 28 days post-randomization. This protocol is reported in line with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines4.
The TOGETHER Trial is an adaptive, multi-arm platform trial with equal allocation of interventions and placebo. The setting for the trial is 10 primary care and emergency department outpatient clinics in the Brazilian state of Minas Gerais.
The primary objective is to determine if each of the IPs reduces: 1) Emergency room visits due to the clinical worsening of COVID-19 (defined as participant remaining under observation for > 6 hours) within 28 days of randomization; 2) Hospitalization due to the progression of COVID-19 (defined as worsening of viral pneumonia) and/or complications within 28 days of randomization.
The secondary objectives are to evaluate, in comparison with placebo, the effect of the IPs on the following parameters:
All-cause, respiratory, and cardiovascular death
Viral clearance and viral load on day 3 and 7 after randomization (conducted the first 150 randomized participants)
Number of days with respiratory symptoms since randomization
Time between the start of treatment until the need for hospitalization/urgent care due to the progression of COVID-19
Rate of all-cause and COVID-specific hospitalizations
Time between the start of treatment and the need for hospitalization for any reason
Quality of life scale and symptoms (PROMIS-10 scale and WHO scale)
Telephone Interview for Cognitive Status (TICS) memory assessment scale on day 28 post randomization
Time from treatment to death (randomization up to 28 days)
Adverse events, adverse reactions to the study medications, and the proportion of participants who are adherent with the medications will also be assessed
Ethical review for this trial follows the Brazilian standard process of CEP/CONEP approval. The trial protocol is first reviewed by the local ethics review board in Brazil, followed by review at the national level by the National Committee for Ethics in Research (CONEP), since the trial is supported by international funding. CONEP approval number: 41174620.0.1001.5120. Research staff members located at the primary care or emergency department clinic where patients first present with symptoms are responsible for obtaining written informed consent. Prospective participants are read the informed consent form which describes trial procedures, potential risks, measures to protect their personal identity, and which parties will have access to their medical information. Ethics certificates from the CEP/CONEP approval process in Brazil are submitted to the Hamilton Integrated Research Ethics Board (HiREB) at McMaster University, which serves as the Ethics Board of Record, for final review and approval. HiREB project number: 13390.
The inclusion criteria are:
1. Patients 18 years and over with the ability to provide free and informed consent;
2. Patients presenting to an outpatient care setting with an acute clinical condition compatible with COVID-19 and symptoms beginning within 07 days from the randomization date;
3. Patients over 18 years and with at least ONE of the following criteria:
a) Age ≥ 50 years (does not need any other risk criteria)
b) Diabetes mellitus requiring oral medication or insulin
c) Systemic arterial hypertension requiring at least 1 oral medication for treatment
d) Known cardiovascular diseases (heart failure, congenital heart disease, valve disease, coronary artery disease, cardiomyopathies being treated, clinically manifested heart disease and with clinical repercussion)
e) Symptomatic lung disease and / or being treated (emphysema, fibrosing diseases)
f) Symptomatic asthma patients requiring chronic use of agents to control symptoms
g) Obesity, defined as BMI> 30 kg / m2 (weight and height information provided by the patient)
h) Transplant patients
i) Patient with stage IV chronic kidney disease or on dialysis
j) Immunosuppressed patients / using corticosteroid therapy (equivalent to a maximum of 10 mg of prednisone per day) and / or immunosuppressive therapy
k) Patients with a history of cancer in the last 55 years or undergoing current cancer treatment
l) Patients with documented fever at screening (>38°C)
m) Patients with at least one of the following symptoms: cough, dyspnea, pleuritic chest pain AND/OR myalgias with limited daily activities (to a maximum of 25% of enrollment)
4. Patients with a positive rapid test for SARS-CoV-2 antigen performed at the time of screening or patients with positive SARS-CoV-2 diagnostic test within 7 days of symptom onset.
5. Willingness to use the proposed investigational treatment and follow the research procedures.
6. Female patients of childbearing potential and male patients with partners of childbearing potential must agree to use adequate methods of contraception during the study and through 90 days after the last dose of study medication.
Participants who already have a positive reverse transcription polymerase chain reaction (RT-PCR) test for SARS-CoV-2 at the time of screening and meet all the inclusion criteria in the survey will not need a new confirmatory test for COVID-19 and can be considered eligible for the randomization / treatment.
Patients who meet any of the following criteria are excluded:
1. Diagnostic examination for SARS-CoV-2 negative associated with acute flu-like symptoms (patient with negative test taken early and becoming positive a few days later is eligible, if he/she is <7 days after the onset of flu-like symptoms);
2. Patients with acute respiratory condition compatible with COVID-19 treated in the primary care and with hospitalization need;
3. Patients with acute respiratory condition due to other causes;
4. Patients who have received at least one dose of vaccination for SARS-CoV-2 >14 days prior to screening;
5. Dyspnea secondary to other acute and chronic respiratory causes or infections (e.g., decompensated chronic obstructive pulmonary disease (COPD), acute bronchitis, pneumonia, primary pulmonary arterial hypertension);
6. Acute flu showing at least ONE of the criteria below:
7. Use of the following medications in the last 14 days:
8. Patients using serotonin reception inhibitors (Donepezil, Sertraline)
9. Pregnant or breastfeeding patients;
10. History of severe ventricular cardiac arrhythmia (ventricular tachycardia, patients with recovered ventricular fibrillation) or long QT syndrome;
11. History of diabetic ketoacidosis or clinical condition that maintains persistent metabolic acidosis;
12. Surgical procedure or use of contrast planned to occur during treatment or up to 05 days after the last dose of the study medication;
13. Current daily and / or uncontrolled alcoholism or drug addiction, what, in the investigator's view, could compromise participation in the study;
14. History of seizures in the last month or uncontrolled seizure;
15. Clinical history of moderate to severe hepatic deficiency or liver cirrhosis or Child-Pugh C classification;
16. Patients with known severe degenerative neurological diseases and / or severe mental illness;
17. Inability of the patient or representative to give informed consent or adhere to the procedures proposed in the protocol;
18. Known hypersensitivity and / or intolerance to IPs, or taking medications contraindicated by IPs;
19. Inability to follow protocol-related procedures
Patients presenting to an outpatient clinic setting with clinical criteria for presumptive diagnosis of COVID-19 who meet the above eligibility criteria are invited to participate in the trial. Nurses, clinicians and health workers will obtain written informed consent from potential trial participants. After obtaining informed consent, research personnel collect demographic information and medical history, and confirm positivity for SARS-CoV-2 using the Abbott Panbio rapid antigen testing for previously undiagnosed patients.
Participants are randomly assigned with equal allocation using a pre-generated randomization list based on block sizes of 8. The block sizes may be increased or decreased depending on the number of active treatment arms. Allocation of participants to treatment arms is uniform across all concurrent interventions as well as placebo. Treatment allocation occurs using a central WhatsApp number where study staff text blocking criteria (e.g. age and co-morbidities) and an unblinded pharmacist replies to the message within 5-10 minutes with the medication letter and randomization number.
Different placebos may be used depending on which IPs are included. For example, if IPs are being administered in both pill format and by injection, participants randomized to the placebo group will be randomly assigned to receive a placebo pill or a placebo injection. If IPs of different duration are being used (e.g. 1 day, 3 days, 10 days, 14 days), participants randomized to the placebo group will be randomized to different placebo durations or regimens.
The randomization is stratified by clinical site and by age (<50 years vs. >=50 years). The randomization sequence for each clinical site is prepared by the unblinded study pharmacist at each participating clinical site. Allocation of treatment assignment is concealed from all other study personnel.
Randomization information is kept confidential by an unblinded statistician. Data are unblinded at the time of the planned interim analyses and at the end of the trial. The trial is quadruple blinded, with participants, research personnel, sponsors, and designees. The Data and Safety Monitoring Committee (DSMC) do not have access to the patient's allocation during review of the interim analysis data, except in the foreseen situations (i.e., decision to stop a treatment arm, termination of the trial, or safety concerns).
The master protocol format and the adaptive design allows the easy addition of different IPs. Ethics approvals are obtained before adding a new IP. Participants are prescribed the IPs and corresponding placebo as indicated in the protocol. An unblinded pharmacist at each clinical site prepares the IP or placebo as per the randomization sequence. The IPs are shipped and stored in a temperature-controlled manner as per the requirements for each IP. Table 1 shows the previous, current (at the time of writing), and planned IPs of investigation in the TOGETHER Trial.
| Investigational Product (IP) | Dose | Dosing schedule | Route of administration |
|---|---|---|---|
| Hydroxychloroquine* | 400mg | Two tablets on Day 1, then 1 tablet for 9 days | Oral |
| Lopinavir/ritonavir* | 200/50mg | Four tablets twice a day on Day 1, then two tablets twice a day for 9 days | Oral |
| Fluvoxamine Maleate | 100mg | One tablet every 12 hours for 10 days | Oral |
| Ivermectin | 400 mcg/kg up to 90kg weight | 3–6, 6mg tablets (weight dependent) every 24 hours for 3 days | Oral |
| Metformin Extended Release | 750mg | One tablet every 12 hours for 10 days | Oral |
| Doxazosin | 2mg | Progressive dosing conditioned on SBP <120 mmHg; 0.5 tablet Day 1–2, 1 tablet Day 3–4, 2 tablets Day 5–7, 3 tablets Day 8–10, 4 tablets Day 11–14 | Oral |
| Pegylated Interferon Lambda | 180 mcg in 0.45 mL | One injection at randomization | Sub-cutaneous injection in lower abdomen |
*Reis G, Moreira Silva E, Medeiros Silva DC, Thabane L, Singh G, Park JJH, Forrest JI, Harari O, Quirino Dos Santos CV, Guimaraes de Almeida APF et al: Effect of Early Treatment With Hydroxychloroquine or Lopinavir and Ritonavir on Risk of Hospitalization Among Patients With COVID-19: The TOGETHER Randomized Clinical Trial. JAMA Netw Open 2021, 4(4):e216468.
Study data are collected on a paper record by the study staff member either in-person at the clinic or by WhatsApp video or voice call with the participant. Data are entered into the IBM electronic case report forms (eCRFs) at each study site. Data quality checks are first performed at the site level, and secondary data checks are performed by central research staff, located at the research coordinating office in the Minas Gerais capital of Belo Horizonte. Weekly meetings are held by Zoom with all study sites to provide feedback on data quality and completeness and continuous training is provided to each site following any changes to the eCRFs or other study procedural amendment. The CRFs can be found as Extended data4.
The primary outcome of the trial is a composite of 1) emergency room visits due to the clinical worsening of COVID-19 (defined as participant remaining under observation for > 6 hours) and 2) hospitalization due to the progression of COVID-19 (defined as worsening of viral pneumonia) and/or complications.
Secondary outcomes include: 1) viral clearance and viral load, 2) time to clinical improvement, defined as the first day on which the participant reports a score of 0 on the WHO Clinical Worsening Scale, 3) number of days with respiratory symptoms since randomization, 4) time to hospitalization for any cause, 5) time to hospitalization due to COVID-19 progression, 6) all-cause, cardiovascular, and respiratory death and time to death from any causes, 7) WHO clinical worsening scale scores over the follow-up period, 8) WHO clinical worsening scale scores during the treatment phase, 9) health-related quality of life (PROMIS global health scale (“Global-10”), and, 10) cognitive status (Telephone Interview for Cognitive Status [TICS]). Adverse events, adverse reactions to the study medications and the proportion of participants who are non-adherent with the study drugs will also be assessed. All secondary outcomes are assessed up to 28 days following randomization.
The study activities for capturing these outcomes at each visit is displayed in Table 2 and Figure 1.
| Screening Visit (D-0) | Baseline and Randomization (1) D-0 | Day 1 | Day 2(4) | Day 3(4) ± 1 day | Day 4 (4) | Day 5(4) | Day 7(4) ± 1 day | Day 10 ± 2 days | Day 14(4) ± 2 days | Day 28(4) ± 3 days | Day 60(4,8,9) or Early Termination ± 5 days | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Informed Consent | X | |||||||||||
| SARS-CoV-2 Rapid Test | X(1) | |||||||||||
| Eligibility Criteria Review | X(2) | |||||||||||
| Pregnancy Test | X(3) | |||||||||||
| Demographics | X(5) | |||||||||||
| Co-morbidities and Risk Factors | X | |||||||||||
| Medical History | X | |||||||||||
| WHO Clinical Worsening Scale | X | X | X | X | X | X | X | X | X | X | X | |
| Exposure to Index Case Information | X | |||||||||||
| Substance Abuse | X | |||||||||||
| PROMIS Global Health Scale | X(6) | X(6) | X(6) | X(6) | ||||||||
| ECG | X | |||||||||||
| Height and Weight | X | |||||||||||
| Nasopharyngeal Swab | X | X | X | |||||||||
| Randomization | X | |||||||||||
| Concomitant Medications | X | X | X | X | X | X | X | X | X | X | X | |
| Investigational Treatment Administration | X(7) | X(7) | X(7) | X(7) | X(7) | X(7) | X(7) | X(7) | ||||
| Hospitalization / Emergency Room Visits | X | X | X | X | X | X | X | X | X | X | ||
| Respiratory Symptoms | X | X | X | X | X | X | X | X | X | X | ||
| Adverse Events | X | X | X | X | X | X | X | X | X | X | ||
| Adverse Drug Reactions | X | X | X | X | X | X | X | X | X | X | ||
| Vaccination Status | X | X | X | |||||||||
| TICS scale - Memory Evaluation | X |
Legend
1. Screening and baseline visit: must be carried out at the same time when attending the outpatient setting. Rapid antigen test for COVID-19 at the screening visit. Day 1 visit should also be conducted on the same day as the screening and baseline visit. After completing the screening visit procedures at the baseline visit and present all inclusion / exclusion criteria, participants should be immediately randomized. The first dose of IP must be administered on the same day of randomization (immediately after randomizing). The study medication will be administered as prescribed. Patients must be observed for 30 minutes after the medication administration.
2. Patients can be included in the trial if they have a COVID-19 diagnosis at baseline visit and have less than 7 days of flu-like symptoms.
3. Only women of childbearing potential and / or potential to become pregnant. Women of childbearing potential must necessarily use contraception during the first 15 days of the trial.
4. Visits through telephone contact, video call, telemedicine are calculated from the randomization date.
5. After signing the Informed Consent Form.
6. Questionnaires must be completed BEFORE any procedures of the proposed visit. Only a person not related to the research can help the patient during the questionnaire. In telephone visits, the patient must respond directly, at the time of contact.
7. Maintain the administration of the IP according to schedule. Discontinue it if adverse events prevent the IP from continuing.
8. Assessment of late complications associated with COVID-19.
9. Unscheduled visits may also be conducted as needed. The clinical outcome data collected at the unscheduled visit should be entered at the next scheduled visit. The treatment period is up to 14 days.
COVID-19=coronavirus disease 2019, SARS-CoV-2=severe acute respiratory syndrome coronavirus 2, WHO=World Health Organisation, ECG=electrocardiogram, TICS=Telephone Interview for Cognitive Status.
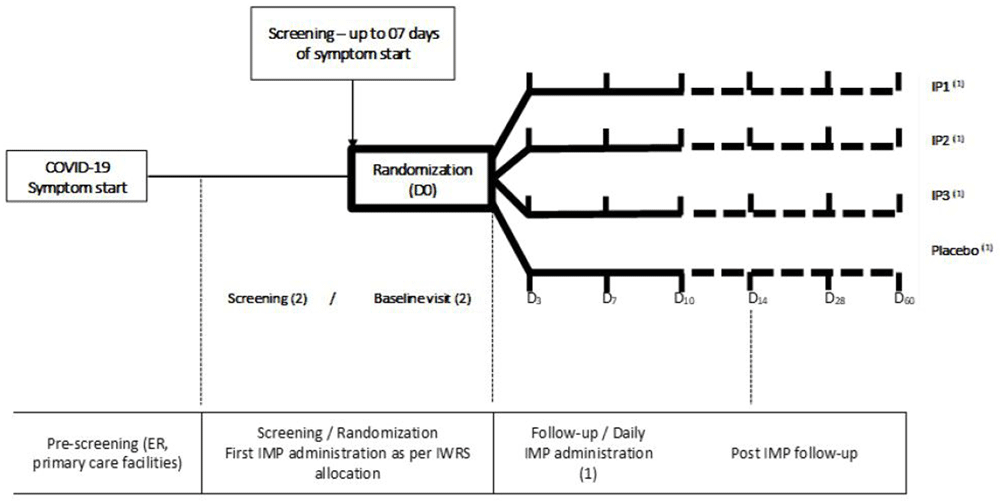
1. Treatment: Investigational Products (IP) IP 1, IP 2, IP 3 in parallel groups for the planned period. Discontinue if significant symptoms or adverse reactions. 2. Screening and randomization (baseline visit) must be performed on the same visit. Ensure that the patient is randomized when at medical care facility. Patient with confirmed SARS-CoV-2 positive test and less than 7 days of symptom onset can be considered for randomization. 3. Subsequent visits: D3, D7, D10, D14, D28, D60 will be carried out by primarily by telephone and/or social media App. Extra visits for safety purposes can be made at any time. Visits D14 and D28 are considered outcome visits as per protocol. D60 is considered post-study visit for monitoring late complications related to COVID-19 and eventual evaluation of late adverse reactions to research drugs and will be carried out by telephone. There is no provision for face-to-face visits in this research in view of the regulatory recommendations issued by the public health authority in the context of the pandemic. 4. Daily contact by phone (not marked above) will be made between Days 1 to 7. Phone contact after D7 will be performed as per protocol. COVID-19=coronavirus disease 2019, SARS-CoV-2=severe acute respiratory syndrome coronavirus 2.
All participants receive standard treatment for COVID-19 as adopted by the health units to which they are linked, as defined by the medical assistant team. All participants will also receive 24-hour telephone contact number which they can call if they have any questions about the trial, if their condition worsens, or if they experience an adverse event. Participants will self-collect nasal swab and saliva for RT-PCR on day 3 and day 7 after randomization. Participants are instructed on this home sample collection and on the logistics for sample retrieval from their residence by study personnel.
The majority of patient evaluations are carried out by telephone contact, social media apps (e.g. WhatsApp), video calls or telemedicine. Face-to-face visits are limited as the virus is highly transmissible. Participants will follow the local health authorities’ guidelines regarding isolation and quarantine requirements, which are generally 14 days from a positive COVID-19 test. Only the day-14 visit will be conducted face-to-face to enable study personnel to collect the medication kits for drug accountability and treatment compliance.
A number of procedures are implemented to maximize participant retention. An informative video recorded by the principal investigator and take-home flyer encourages patients to adhere to study procedures and complete the trial. Participants are also sent occasional notification reminders on WhatsApp encouraging trial participation and follow-up appointments by WhatsApp are conducted in an effort to minimize travel days to the clinic.
By Brazilian regulations, we provide medical care to all patients throughout their participation (2 months). For longer term events, the trial insurance covers any study related adverse event for a period of three years post-randomization. Auditing of the trial occurs at the central level with 50–60% Source Data Verification (SDV).
A Steering Committee and an independent DSMC have been established. The Steering Committee oversees the study to ensure scientific integrity and routinely assess emerging evidence to recommend interventions of interest for the trial. The DSMC oversees the safety of the research participants and reviews the results of each interim analysis and final analysis and makes recommendations on stopping or continuing each IP. Events of special interest flagged by the DSMC are followed-up by study physicians.
Members of the Data Safety and Monitoring Committee include Dr William Cameron of University of Ottawa (Canada), Dr James Orbinski of York University (Canada), Dr Sonal Singh of University of Massachusetts (USA), Dr. Kristian Thorlund of McMaster University (Canada) and Dr. Jonas Haggstrom of Cytel Inc. (Sweden).
Our trial applies sample size reassessment according to observed events. The trial is a platform adaptive trial design with three planned interim analyses at approximately 25%, 50%, and 75% of the total required sample size. The initial sample size calculation is based on the test for the hypothesis that each of the IPs will be better than placebo in reducing the risk of hospitalization and emergency room care at least 6 hours in duration due to complications directly related to COVID-19. The sample size of 681 patients per arm was chosen for each experimental group to achieve 80% power with 0.05 two-sided Type 1 error for a pairwise comparison against the control to detect minimum treatment efficacy defined by 37.5% relative risk reduction (RRR) of preventing hospitalization assuming a control event rate (CER) of 15%. The sample size calculation will be revised based on the outcomes that occurred during the interim analyses. Blind analysis of outcomes with simulations will be conducted to limit type I errors within the 5% tolerance range (97.5% or greater probability of superiority over the control group). Individual treatment arms can be stopped if there are no acceptable projections of benefit at the expense of futility.
Interim efficacy analyses are scheduled. Assuming a uniform prior assigned to the different event rates, a total sample size of 681 patients per arm, a CER of 15%, and a RRR equals to 37.5%, an interim analysis will be performed after observing approximately 25%, 50% and 75% of the maximum number of patient outcomes, as well as at the trial completion. The posterior efficacy threshold to stop for superiority is 97.6% and the futility threshold is 20%, 40% and 60% at the respective interim analyses. Intervention arms(s) showing a posterior probability of efficacy crossing either boundary, will be stopped for either reason. These superiority and futility thresholds were determined based on 200,000 simulation run ins wherein different values of the RRR were considered (0%, 20%, and 37.5%). A description of this interim analysis in an event-based Bayesian adaptive trial and accompanying illustrating example can be found in the appendix of this document.
When other data from other relevant studies become available, we will use Empirical Bayes meta-analysis5 to borrow information from the treatment effects or safety signals emerging from these studies. This is effectively a random effect Bayesian model that results in simultaneous shrinkage of the treatment effect or safety estimates reported in the various studies toward the meta-analysis estimate, while still providing standalone estimates. Schoenfeld et al. have shown6 that this approach is, in some ways, equivalent to the power prior approach of Chen and Ibrahim7, whereby historical studies are assigned fractional weight(s) whose magnitudes correspond to the consistency of their data with that of the study they are thought to inform. The analyses incorporating external evidence will be presented to the DSMC as secondary findings to consider but will not alone trigger a recommendation for a trial adaptation.
A detailed description of the TOGETHER Trial statistical analysis plan can be found in the Extended data4.
In brief, the efficacy of each intervention will be analyzed in terms of its posterior efficacy with respect to placebo, using the Bayesian paradigm, while calibrating the decision boundaries to meet the type I error rate requirements. We will adopt an intention-to-treat principle to analyze all results. Multiple imputation will be employed where statistical models require adjustment for baseline covariates with up to 20% missing values. No multiple imputation of outcomes will be performed.
We will use Bayesian inference for dichotomous outcomes, adjusting for covariates when necessary. Similarly, we will validate the proportional hazards assumption by visually inspecting Kaplan-Meier and log-negative-log of survival plots and fit Cox model for time-to-event outcomes. Secondary outcomes, such as viral clearance will be modelled using a longitudinal logistic model with a subject random effect, using the PCR test result over time as our dependent variable.
Subgroup analyses
We will perform subgroup analyses to assess the consistency of effects in four patient subgroups:
We hypothesize that younger patients will benefit more than older patients, women will benefit more than men, patients with an earlier diagnosis will benefit more than those with a later diagnosis, and patients without the clinical co-morbidities described above will benefit more than those with these co-morbidities. All the subgroup hypotheses are based on data emerging from other countries, indicating the differential impact of COVID-19 by age, sex and the existence of clinical comorbidities at baseline conditions.
Data from the IBM eCRFs are securely sent by File Transfer Protocol (FTP) to the statistical team in SAS format. SAS v9.4 is used to convert raw data into an analytic dataset applying CDISC standards. Analyses are conducted using R v4.0.3. Results will be reported following the CONSORT guidelines.
The funder of this trial had no role in study design, data collection, decision to publish, or preparation of the manuscript.
The final trial dataset will be accessible by written request to the study principal investigators (G Reis or EJ Mills). There are no contractual agreements to limit access to final trial data. All data collected by the TOGETHER Trial will be shared with the International COVID-19 Data Alliance. Access to these data through the ICODA Workbench will follow the standard operating procedures developed by the ICODA working group.
Findings will be disseminated in several ways. All investigations of IPs vs. placebo will be submitted to an appropriate, peer-reviewed scientific journal. Lay summaries of findings will be made available on the TOGETHER Trial website (togethertrial.com). The investigative team is also connected to the WHO COVID-19 guidelines committee, where trial findings will help inform global clinical guidance.
The TOGETHER Trial has recruited more than 3000 patients to date. The trial has previously evaluated the effect of hydroxychloroquine or lopinavir/ritonavir on risk of hospitalization8. An arm evaluating metformin vs. placebo was stopped early by the DSMC for futility. Other arms evaluating ivermectin and fluvoxamine are continuing. Future planned evaluations will include doxazosin and pegylated interferon lambda. The IPs of investigation in the TOGETHER Trial and their study status at the time of writing is further described in Table 1.
Our TOGETHER trial is innovative in a number of ways. First, from a clinical perspective, we are examining the use of drugs that would be widely available and accessible if proven effective and safe for the treatment of COVID-19. Second, our trial uses a new methodological approach adaptable to both internal accumulating data, as is common in platform trials, as well as incorporate external trial evidence that may be unplanned at the time of initial study launch.
Currently, there are no effective approved therapeutic interventions approved for the early treatment of SARS-CoV-21,9–11. Proposed therapies for SARS-CoV-2 are based on previous clinical experience directed against SARS-CoV-1 and Middle East respiratory syndrome (MERS)12. These therapeutic modalities consisted of viral methyl transferase inhibitors, protease inhibitors, interferon, inhibitors of viral ribonucleic acid (RNA) synthesis as well as anti-inflammatory drugs. For the treatment of COVID-19, there has been much promise and excitement for repurposing drugs that have similar targets described for SARS-CoV-1 and MERS1,13. The current use of repurposed drugs for COVID-19 treatment offers several key advantages as these medicines have been proven safe, their pharmacokinetics are well understood, and optimal dosages are standardized. Although hydroxychloroquine is ineffective for the treatment of COVID-19 among hospitalized adults14, other repurposed drugs have already shown promise against COVID-19 disease at the later stages of disease. Both dexamethasone and tociluzimab appear to significantly increase survival according to findings from the UK RECOVERY trials15. Furthermore, other new molecules such as remdesivir and monoclonal antibodies have had inconsistent findings16,17. Unfortunately, well-designed studies on asymptomatic or mild, or pediatric cases of COVID-19 are lacking. Neither hydroxychloroquine nor lopinavir-ritonavir showed any significant benefit for decreasing COVID-19-associated hospitalization or other secondary clinical outcomes in early symptomatic COVID-19 patients8. In a preliminary study of adult outpatients with early COVID-19, patients treated with fluvoxamine, compared with placebo, had a lower likelihood of clinical deterioration over 15 days18. In another recent trial of favipiravir, an RNA-dependent RNA polymerase inhibitor, also did not show any statistically significant benefit in term of mortality in the general group of patients with mild to moderate COVID-1919. It was suggested that the use of antivirals in symptomatic patients is too late and would explain their low efficacy in the clinical setting. A number of clinical trials (NCT04426695, NCT04425629, NCT04479631) have now been initiated to assess safety, tolerability, and efficacy of SARS-CoV-2 neutralizing monoclonal antibodies (nMAbs) using either a prophylactic or therapeutic approach20–22. In addition, potent human monoclonal antibodies against SARS-CoV-2 have been isolated from COVID-19 convalescent patients which could provide another layer of therapeutic options against the disease23. Thus, by blocking acute virus replication, early nMAb intervention would potentially induce a better clinical outcome against COVID-19.
Our study has several limitations. Perhaps the greatest limitation of our study is that the administrative stages of conducting a trial, from protocol development and ethics review to obtaining study drug and creating electronic case report forms are all reliant on the local infrastructure and norms of study conduct in those settings. Our adaptive elements of the trial complicate what is understood by some agencies and push-back from approval bodies has previously delayed enrolment. The rapid change in the scientific interest or confidence in interventions means that an application submitted to a funding agency or ethics committee, may, by the time it is reviewed, have changed dramatically. Strengths of our study include the adaptive nature of the study to change arms by dropping or adding arms as the data, both internal and external. Our design permits outside data, from either trial we already collaborate with or trials with emerging data we learn of as we are conducting our trial. Similarly, outside evidence in the form of completed trials, may provide sufficiently compelling evidence to change the direction of our trial or change outcomes and interpretation of trial findings.
Our design is adaptive and also Bayesian in its learning structure and analysis. We refer to this as a learning structure as emerging data from our own trial will, almost certainly be influenced from data we were unaware of at the study outset. We are already learning of similar trials examining similar interventions (in at least one arm) where the population inclusion criteria and the outcomes can be harmonized with our dataset. Similarly, we may find out about completed trials that have convincing evidence that mandates a change in our trial. For example, if a large study with a similar population and outcome found overwhelming evidence of a treatment effect (whether that is harm, futility, or benefit), we may examine our data to confirm that the direction of treatment effect is similar. This may take the form of matching the population using a strategy such as propensity scoring or combining in a meta-analysis.
Results from this trial will help identify repurposed therapeutics for COVID-19 that can easily be scaled in low- and middle-income settings. The novel methodological extension of the platform adaptive design to dynamically incorporate external evidence will be the first of its kind and may prove highly valuable for all COVID-19 trials and trials for other indications going forward.
Open Science Framework: A multi-center, adaptive, randomized, platform trial to evaluate the effect of repurposed medicines in outpatients with early coronavirus disease 2019 (COVID-19) and high-risk for complications: the TOGETHER master trial. https://doi.org/10.17605/OSF.IO/EG37X4.
This project contains the following extended data:
Open Science Framework: SPIRIT checklist for ‘A multi-center, adaptive, randomized, platform trial to evaluate the effect of repurposed medicines in outpatients with early coronavirus disease 2019 (COVID-19) and high-risk for complications: the TOGETHER master trial protocol’. https://doi.org/10.17605/OSF.IO/EG37X4.
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| Gates Open Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with Gates Open Research
Already registered? Sign in
If you are a previous or current Gates grant holder, sign up for information about developments, publishing and publications from Gates Open Research.
We'll keep you updated on any major new updates to Gates Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)