Keywords
Bacterial endosymbionts, sub-Saharan Africa, cassava, smallholder farmers, NusG, next generation sequencing
Bacterial endosymbionts, sub-Saharan Africa, cassava, smallholder farmers, NusG, next generation sequencing
Members of the whitefly Bemisia tabaci (Hemiptera: Aleyrodidae) species complex are classified as the world’s most devastating insect pests. There are 34 species globally1 and the various species in the complex are morphologically identical. They transmit over 100 plant viruses2,3, become insecticide resistant4, and ultimately cause billions of dollars in damage annually for farmers. The adult whiteflies are promiscuous feeders, and will move between viral infected crops and native weeds that act as viral inoculum ‘sources’, and deposit viruses to alternative crops that act as viral ‘sinks’ while feeding.
The crop of importance for this study was cassava (Manihot esculenta). Cassava supports approximately 800 million people in over 105 countries as a source of food and nutritional security, especially within rural smallholder farming communities5. Cassava production in Sub Saharan Africa (SSA), especially the East Africa region, is hampered by both DNA and RNA transmitted viruses.
Whitefly-transmitted viruses cause cassava mosaic disease (CMD) leading to 28-40% crop losses with estimated economic losses of up to $2.7 billion dollars per year in SSA6. The CMD pandemics in East Africa, and across other cassava producing areas in SSA, were correlated with B. tabaci outbreaks7. Relevant to this study are two RNA Potyviruses: Cassava Brown Streak Virus (CBSV) and the Uganda Cassava Brown Streak Virus (UCBSV), both devastating cassava in East Africa. Bemisia tabaci species have been hypothesized to transmit these RNA viruses with limited transmission efficiency8–10. Recent studies have shown that there are multiple species of these viruses11, which further strengthens the need to obtain data from individual whiteflies as pooled samples could contain different species with different virus composition and transmission efficiency. In addition, CBSV has been shown to have a higher rate of evolution than UCBSV12 increasing the urgency of understanding the role played by the different whitefly species in the system.
Viral-vector interactions within B. tabaci are further influenced by bacterial endosymbionts forming a tripartite interaction. B. tabaci has one of the highest numbers of endosymbiont bacterial infections with eight different vertically transmitted bacteria reported13–16. They are classified into two categories; primary (P) and secondary (S) endosymbionts, many of which are in specialised cells called bacteriocytes, while a few are also found scattered throughout the whitefly body. A single obligate P-symbiont P. aleyrodidarum is systematically found in all B. tabaci individuals. Portiera has a long co-evolutionary history with all members of the Aleyrodinae subfamily15. In this study, we further explore genes within the P. aleyrodidarum retrieved from individual whitefly transcriptomes, including the transcription termination/antitermination protein NusG. NusG is a highly conserved protein regulator that suppresses RNA polymerase, pausing and increasing the elongation rate17,18. However, its importance within gene regulation is species specific; in Staphylococcus aureus it is dispensable19,20.
The S-endosymbionts are not systematically associated with hosts, and their contribution is not essential to the survival and reproduction. Seven facultative S-endosymbionts, Wolbachia, Cardinium, Rickettsia, Arsenophonus, Hamiltonella defensa and Fritschea bemisae have been detected in various B. tabaci populations13,21–24. The presence of S-endosymbionts can influence key biological parameters of the host. Hamiltonella and Rickettsia facilitate plant virus transmission with increased acquisition and retention by whiteflies22. This is done by protection and safe transit of virions in the haemolymph of insects through chaperonins (GroEL) and protein complexes that aid in protein folding and repair mechanisms19.
The advent of next generation sequencing (NGS) and specifically transcriptome sequencing has allowed the unmasking of this tripartite relationship of vector-viral-microbiota within insects24–28. Furthermore, NGS provides an opportunity to better understand the co-evolution of B. tabaci and its bacterial endosymbionts26. The endosymbionts have been implicated in influencing species complex formation in B. tabaci through conducting sweeps on the mitochondrial genome27. Applying transcriptome sequencing is essential to reveal the endosymbionts and their effects on the mitogenome of B. tabaci, and predict potential hot spots for changes that are endosymbiont induced.
Several studies have explored the interaction between whitefly and endosymbionts29,30 and have resulted in the identification of candidate genes that maintain the relationship31,32. This has been explored as a source of potential RNAi pesticide control targets29,32,33. RNAi-based pest control measures also provide opportunities to identify species-specific genes for target gene sequences for knock-down. However, to date all transcriptome sequencing has involved pooled samples, obtained through rearing several generations of isolines of a single species to ensure high quantities of RNA for subsequent sequencing. This remains a major bottle neck in particular within arthropoda, where collected samples are limited due to small morphological sizes34,35. In addition, the development of isolines is time consuming and often has colonies dying off mainly due to inbreeding depression33.
It is against this background that we sought to develop a method for single whitefly transcriptomes to understand the virus diversity within different whitefly species. We did not detect viral reads, probably an indication that the sampled whitefly was not carrying any viruses, but as proof of concept of the method, we validated the utility of the data generated by retrieving the microbiota P-endosymbionts and S-endosymbionts that have previously been characterised within B. tabaci. In this study we report the endosymbionts present within field-collected individual African whiteflies, as well as characterisation and evolution of the NusG genes present within the P-endosymbionts.
In this study, we sampled whiteflies in Uganda and Tanzania from cassava (Manihot esculenta) fields. In Uganda, fresh adult whiteflies were collected from cassava fields at the National Crops Resources Research Institute (NaCRRI), Namulonge, Wakiso district, which is located in central Uganda at 32°36’E and 0°31’N, and 1134 meters above sea level. The whiteflies obtained from Tanzania were collected on cassava in a countrywide survey conducted in 2013. The samples: WF2 (Uganda) and WF1, WF2a, and WF2b (Tanzania) used in this study were collected on CBSD-symptomatic cassava plants. In all the cases, the whitefly samples were kept in 70% ethanol in Eppendorf tubes until laboratory analysis. The whitefly samples were used for a two-fold function; firstly, to optimise a single whitefly RNA extraction protocol and secondly, to unmask RNA viruses and endosymbionts within B. tabaci as a proof of concept. In addition, data obtained from Nextera – DNA library prep from a Brazilian sample (156_NW2) was also used in this study. The whitefly was collected from a New World 2 colony in Brazil on Euphorbia heterophylla and kept in 70% ethanol in Eppendorf tubes until laboratory analysis.
RNA extraction was carried out using the ARCTURUS® PicoPure® kit (Arcturus, CA, USA), which is designed for fixed paraffin-embedded (FFPE) tissue samples. Briefly, 30 µl of extraction buffer was added to an RNase-free micro centrifuge tube containing a single whitefly and ground using a sterile plastic pestle. To the cell extract an equal volume of 70% ethanol was added. To bind the RNA onto the column, the RNA purification columns were spun for two minutes at 100 x g and immediately followed by centrifugation at 16,000 x g for 30 seconds. The purification columns were then subjected to two washing steps using wash buffer 1 and 2 (ethyl alcohol). The purification column was transferred to a fresh RNase-free 0.5 ml micro centrifuge tube, with 30 µl of RNAse-free water added to elute the RNA. The column was incubated at room temperature for five minutes, and subsequently spun for one minute at 1,000 x g, followed by 16,000 x g for one minute. The eluted RNA was returned into the column and re-extracted to increase the concentration. Extracted RNA was treated with DNase using the TURBO DNA free kit, as described by the manufacturer (Ambion, Life Technologies, CA, USA). Concentration of RNA was done in a vacuum centrifuge (Eppendorf, Germany) at room temperature for 1 hour, the pellet was suspended in 15 µl of RNase-free water and stored at -80°C awaiting analysis. RNA was quantified, and the quality and integrity assessed using the 2100 Bioanalyzer (Agilent Technologies, CA, USA). Dilutions of up to x10 were made for each sample prior to analysis in the bioanalyzer.
Total RNA from each individual whitefly sample was used for cDNA library preparation using the Illumina TruSeq Stranded Total RNA Preparation kit as described by the manufacturer (Illumina, CA, USA). Subsequently, sequencing was carried out using the HiSeq2000 (Illumina) on the rapid run mode generating 2 x 50 bp paired-end reads. Base calling, quality assessment and image analysis were conducted using the HiSeq control software v1.4.8 and Real Time Analysis v1.18.61 at the Australian Genome Research Facility (Perth, Australia).
Assembly of RNA transcripts: Raw RNA-Seq reads were trimmed using Trimmomatic. The trimmed reads were used for de novo assembly using Trinity34 with the following parameters: time -p srun --export=all -n 1 -c ${NUM_THREADS} Trinity --seqType fq --max_memory 30G --left 2_1.fastq --right 2_2.fastq --SS_lib_type RF --CPU ${NUM_THREADS} --trimmomatic --cleanup --min_contig_length 1000 -output _trinity min_glue = 1, V = 10, edge-thr = 0.05, min_kmer_cov = 2, path_reinforcement_distance = 150, and group pairs distance = 500.
BLAST analysis of transcripts and annotation: BLAST searches of the transcripts under study were carried out on the NCBI non-redundant nucleotide database using the default cut-off on the Magnus Supercomputer at the Pawsey Supercomputer Centre Western Australia. Transcripts identical to known bacterial endosymbionts were identified and the number of genes from each identified endosymbiont bacteria determined.
Phylogenetic analysis of whitefly mitochondrial cytochrome oxidase I (COI): The phylogenetic relationship of mitochondrial cytochrome oxidase I (mtCOI) of the whitefly samples in this study were inferred using a Bayesian phylogenetic method implemented in MrBayes \ (version 3.2.2)35. The optimal substitution model was selected using Akaike Information Criteria (AIC) implemented in the Jmodel test 236.
Sequence alignment and phylogenetic analysis of NusG gene in P. aleyrodidarum across B. tabaci species: Sequence alignment of the NusG gene from the P-endosymbiont P. aleyrodidarum from the SSA1 B. tabaci in this study was compared with another B. tabaci species, Trialeurodes vaporariorum and Alerodicus dispersus using MAFFT (version 7.017)37. The Jmodel version 236 was used to search for phylogenetic models with the Akaike information criterion selecting the optimal that was to be implemented in MrBayes 3.2.2. MrBayes run was carried out using the command: “lset nst=6 rates=gamma” for 50 million generations, with trees sampled every 1000 generations. In each of the runs, the first 25% (2,500) trees were discarded as burn in.
The structures for Portiera aleyrodidarum BT and B. tabaci SSA1 whitefly were predicted using Phyre238 with 100% confidence and compared to known structures of NusG from other bacterial species. All models were prepared using Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4).
In this study, we sampled four individual adult B. tabaci from cassava fields in Uganda (WF2) and Tanzania (WF1, WF2a, WF2b). Total RNA from single whitefly yielded high quality RNA with concentrations ranging from 69 ng to 244 ng that were used for library preparation and subsequent sequencing with Illumina Hiseq 2000 on a rapid run mode. The number of raw reads generated from each single whitefly ranged between 39,343,141 and 42,928,131 (Table 1). After trimming, the reads were assembled using Trinity resulting in 65,550 to 162,487 transcripts across the four SSA1 B. tabaci individuals (Table 1).
Comparison of the diversity of bacterial endosymbionts across individual whitefly transcripts was conducted with BLASTn searches on the non-redundant nucleotide database and by identifying the number of genes from each bacterial endosymbiont (Supplementary Table 1). We identified five main endosymbionts including: P. aleyrodidarum the primary endosymbionts and four secondary endosymbionts: Arsenophonus, Wolbachia, Rickettsia sp, and Cardinium spp (Table 2). P. aleyrodidarum predominated all four SSA1 B. tabaci study samples with incidences of 74.8%, 71.2%, 54.1% and 58.5% for WF1, WF2, WF2a and WF2b, respectively. This was followed by Arsenophonus, Wolbachia, Rickettsia sp, and Cardinium spp, which occurred at an average of 18.0%, 5.9%, 1.6% and <1%, respectively across all four study samples.
B. tabaci is recognized as a species complex of 34 species based on the mitochondrion cytochrome oxidase I1,39,40. We therefore used cytochrome oxidase I (COI) transcripts of the four individual whitefly to ascertain B. tabaci species status and their phylogenetic relation using reference B. tabaci COI GenBank sequences found at www.whiteflybase.org. All four COI sequences clustered within Sub Saharan Africa clade 1 (SSA1) species (data not shown).
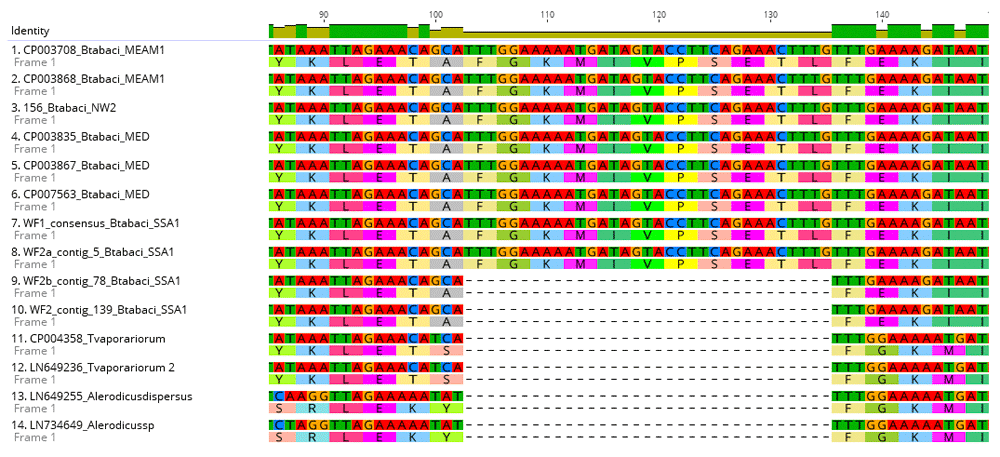
Nucleotide and amino acid sequence alignments of the NusG in P. aleyrodidarum were conducted for several whitefly species including: B. tabaci (SSA1, Mediterranean (MED) and Middle East Asia Minor 1 (MEAM1) New World 2 (NW2), T. vaporariorum (Greenhouse whitefly) and Alerodicus dispersus. The alignment identified 11 missing amino acids in the NusG sequences for the SSA1 B. tabaci samples: WF2 and WF2b, T. vaporariorum (Greenhouse whitefly) and Alerodicus disperses. However, all 11 amino acids were present in samples WF1 and WF2a, MED, MEAM1 and NW2 (Figure 1). Bayesian phylogenetic relationships of the NusG sequences of P. aleyrodidarum for the different whitefly species clustered all four SSA1 B. tabaci (WF1, WF2, WF2a and WF2b) within a single clade together with ancestral B. tabaci from GenBank (Figure 2). The SSA1 clade was supported by posterior probabilities of 1 with T. vaporariorum and Alerodicus, which formed clades at the base of the phylogenetic tree (Figure 2).

Structures of the NusG protein sequence of the primary endosymbiont P. aleyrodidarum in the four SSA1 B. tabaci samples were predicated using Phyre2 with 100% confidence, and compared to known structures of NusG from other bacterial species including (Shigella flexneri, Thermus thermophiles, and Aquifex aeolicus; (PDB entries 2KO6, 1NZ8 and 1M1H, respectively) and Spt4/5 from yeast (Saccharomyces cerevisiae; PDB entry 2EXU)19,41,42. The 11-residue deletion was found in a loop region that is variable in length and structure across bacterial species, but is absent from archaeal and eukaryotic species (Figure 3 and Figure 4A). The effect of the deletion appears to shorten the loop in NusG from the African whiteflies (WF2 and WF2b). A model of bacterial RNA polymerase (orange surface representation; PDB entry 2O5I) bound to the N-terminal domain of the T. thermophiles NusG shows that the loop region is not involved in the interaction between NusG and RNA polymerase (Figure 4B).
Structure analysis of NusG from P. aleyrodidarum in B. tabaci and other endosymbionts A. Phyre2 based structure prediction of NusG from Candidatus Portiera aleyrodidarum in B. tabaci SSAI whitefly and comparisons to the structures of NusG from other bacterial species as indicated and of Spt4/5 from yeast. NusG is coloured in grey, the loop region in magenta and the 11-residue deletion is shown in green in the C. Portiera aleyrodidarum structure. B. A model of bacterial RNA polymerase (orange surface representation) bound to the N-terminal domain of the T. thermophiles NusG (grey cartoon representation).
In this study, we optimised a single whitefly RNA extraction method for field-collected samples. We subsequently successfully conducted transcriptome sequencing on individual Sub-Saharan Africa 1 (SSA1) B. tabaci, revealing unique genetic diversity in the bacterial endosymbionts as proof of concept. This is the first time a single whitefly transcriptome has been produced.
We report the presence of the primary endosymbionts P. aleyrodidarum and several secondary endosymbionts within SSA1 transcriptome. Furthermore, P. aleyrodidarum in SSA1 B. tabaci was observed to have a deletion of 11 amino acids on the NusG gene that is associated with cellular transcriptional processes within another bacteria species. On the other hand, P. aleyrodidarum from NW2, MED and SSA1 (WF2a, WF1) B. tabaci species did not have this deletion (Figure 1). The deleted 11 amino acids were identified in a loop region of the N-terminal domain of NusG protein, resulting in a shortened loop in the SSA1 WF2b sample. This loop region has high variability in both structure and length across bacterial species, and is absent from archaea and eukaryotic species.
NusG is highly conserved and a major regulator of transcription elongation. It has been shown to directly interact with RNA polymerase to regulate transcriptional pausing and rho-dependent termination19,20,43,44. Structural modelling of NusG bound to RNA polymerase indicated that the shortened loop region seen in the WF2b sample is unlikely to affect this interaction. Rho-dependant termination has been attributed to the C-terminal (KOW) domain region of NusG, therefore a shortening of the loop region in the N-terminal domain is also unlikely to affect transcription termination. Yet, there has been no function attributed to this loop region of NusG, and thus the effect of variability in this region across species is unknown. However, the deletion could represent the results of evolutionary species divergence. Further sequencing of the gene is required across the B. tabaci species complex to gain further understanding of the diversity.
The sequencing of the whitefly transcriptome is crucial in understanding whitefly-microbiota-viral dynamics and thus circumventing the bottlenecks posed in sequencing the whitefly genome. The genome of whitefly is highly heterozygous43. Assembling of heterozygous genomes is complex due to the de Bruijn graph structures predominantly used44. To deal with the heterozygosity, previous studies have employed inbred lines, obtained from rearing a high number of whitefly isolines34,45. However, rearing whitefly isolines is time consuming and often colonies may suffer contaminations, leading to collapse and failure to raise the high numbers required for transcriptome sequencing.
We optimised the ARCTURUS® PicoPure® kit (Arcturus, CA, USA) protocol for individual whitefly RNA extraction with the dual aim of determining if we could obtain sufficient quantities of RNA from a single whitefly for transcriptome analysis and secondly, determine whether the optimised method would reveal whitefly microbiota as proof of concept. Using our method, the quantities of RNA obtained from field-collected single whitefly samples were sufficient for library preparation and subsequent transcriptome sequencing. Across all transcriptomes over 30M reads were obtained. The amount of transcripts were comparable to those reported in other arthropoda studies from field collections32. However, we did not observe any difference in assembly qualities32; probably due to the fact that our field-collected samples had degraded RNA based on RIN, and thus direct comparison with32 was inappropriate.
Degraded insect specimen have been used successfully in previous studies46. This is significant, considering that the majority of insect specimens are usually collected under field conditions and stored in ethanol with different concentrations ranging from 70 to 100%47–49 rendering the samples liable to degradation. However, to ensure good keeping of insect specimen to be used for mRNA and total RNA isolation in molecular studies, and other downstream applications such as histology and immunocytochemistry, it is advisable to collect the samples in an RNA stabilizing solution such as RNAlater. The solution stabilizes and protects cellular RNA in intact, unfrozen tissue, and cell samples without jeopardizing the quality, or quantity of RNA obtained after subsequent RNA isolation. The success of the method provided an opportunity to unmask vector-microbiota-viral dynamics in individual whiteflies in our study, and will be useful for similar studies on other small organisms.
In this study, we identified bacterial endosymbionts (Table 2) that were comparable to those previously reported in B. tabaci50 and more specifically SSA1 on cassava24,38,51. Secondary endosymbionts have been implicated with different roles within B. tabaci. Rickettsia has been adversely reported across putative B. tabaci species, including the Eastern African region24,52. This endosymbiont has been associated with influencing thermo tolerance in B. tabaci species53. Rickettsia has also been associated with altering the reproductive system of B. tabaci, and within the females. This has been attributed to increasing fecundity, greater survival, host reproduction manipulation and the production of a higher proportion of daughters all of which increase the impact of virus54. Arsenophonus, Wolbachia Arsenophonus and Cardinium spp have been detected within MED and MEAM1 Bemisia species13,53. In addition, 51 and 21 reported Arsenophonus within SSA1 B. tabaci in Eastern Africa that were collected on cassava. These endosymbionts have been associated with several deleterious functions within B. tabaci that include manipulating female-male host ratio through feminizing genetic males, coupled with male killing54,55.
Within the context of SSA agricultural systems, the role of endosymbionts in influencing B. tabaci viral transmission is important. Losses attributed to B. tabaci transmitted viruses within different crops are estimated to be in billions of US dollars47. Bacterial endosymbionts have been associated with influencing viral acquisition, transmission and retention, such as in tomato leaf curl virus25,56. Thus, better understanding of the diversity of the endosymbionts provides additional evidence on which members of B. tabaci species complex more proficiently transmit viruses, and thus the need for concerted efforts towards the whitefly eradication.
Our study provides a proof of concept that single whitefly RNA extraction and transcriptome sequencing is possible and the method is optimised and applicable to a range of small insect transcriptome studies. It is particularly useful in studies that wish to explore vector-microbiota-viral dynamics at individual insect level rather than pooling of insects. It is useful where genetic material is both limited, as well as of low quality, which is applicable to most agriculture field collections. In addition, the single whitefly transcriptome technique described in this study offers new opportunities to understand the biology, and relative economic importance, of the several whitefly species occurring in ecosystems within which food is produced in Sub-Saharan Africa, and will enable the efficient development and deployment of sustainable pest and disease management strategies to ensure food security in the developing countries.
The datasets used and/or analyzed during the current study are available from GenBank:
SRR5110306, SRR5110307, SRR5109958, KY548924, MG680297.
| Views | Downloads | |
|---|---|---|
| Gates Open Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Register with Gates Open Research
Already registered? Sign in
If you are a previous or current Gates grant holder, sign up for information about developments, publishing and publications from Gates Open Research.
We'll keep you updated on any major new updates to Gates Open Research
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)